
티스토리 뷰
< NGS 데이터 분석 >
- 일반적으로 환자의 혈액 등 샘플을 채취하여 기계에 넣고 sequence를 읽어내는 과정을 Sequencing Base Calling이라고 함. 긴 서열을 쪼개서 반복하여 읽은 각 정보를 read라고 하고, 이를 조합하여 원 서열 정보를 알아냄
- Base Calling이 끝난 데이터를 reference sequence와 비교하여 어느 위치에 변이가 있나를 알아내는 과정이 variant calling임
- variant calling 정보를 통계적으로 처리하여 최종 변이 정보를 저장한 파일이 vcf 파일

- Alignment Variant Calling
Base calling을 완료하면 FASTA 혹은 FASTAQ 파일이 만들어짐.
이 데이터를 SAM -> BAM -> VCF 파일로 처리하는 과정이 alignment variant calling이라고 할 수 있음
1) Trimming FASTAQ for QC
sudo apt install sickle
sickle pe -t sanger -f /data/QC18010001_1.fastq.gz -r /data/QC18010001_2.fastq.gz \
-o /data/QC18010001_1_trim.fastq -p /data/QC18010001_2_trim.fastq \
-s single.fasq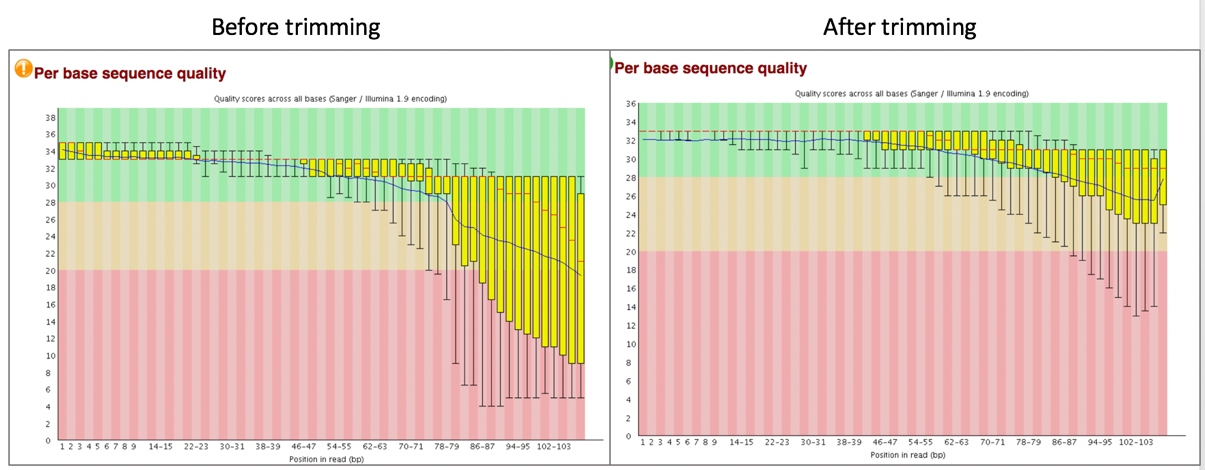
- Trim을 할 수 있는 mutect2나 sickle을 다운 받음
- fastQC를 이용하여 trim된 데이터를 시각화 할 수 있음
녹색 부분은 normal, 노란색은 reasonable, 붉은색은 bad 영역이라고 볼 수 있음
trim 진행 후 read quality가 reasonable 이상 영역으로 정돈된 것을 확인할 수 있음
2. Indexing
- reference 파일과 sample 파일의 위치를 맞추기 위한 indexing 처리가 필요함
- bwa를 이용하여 처리할 수 있음
bwa-0.7.17/bwa index -a bwtsw /ref/hg38/Homo_sapiens_assembly38.fasta #ref. 파일
# trimed sample 파일의 ref에 대한 indexing
bwa-0.7.17/bwa aln /ref/Homo_sapiens_assembly38.fasta \
QC18010001_1_trim.fastq > QC18010001_1_trim.fastq.sai
3. Alignment
- indexing을 통해 위치가 확인되면 그 위치에서의 염기서열을 비교 확인할 수 있는 alignment 과정을 거쳐
확인된 정보를 sai > sam 파일에 저장하게 됨
- 파일의 용량을 줄이기 위해 같은 정보를 binary로 저장한 BAM 파일로 변환
bwa-0.7.17/bwa sampe /ref/Homo_sapiens_assembly38.fasta \
QC18010001_1_trim.fastq.sai QC18010001_2_trim.fastq.sai\
QC18010001_1_trim.fastq QC18010001_2_trim.fastq > QC18010001.sam
# samtools를 이용하여 bam 파일로 변환
samtools view -bS -q 23 QC18010001.sam > QC18010001.bam
4. BAM file
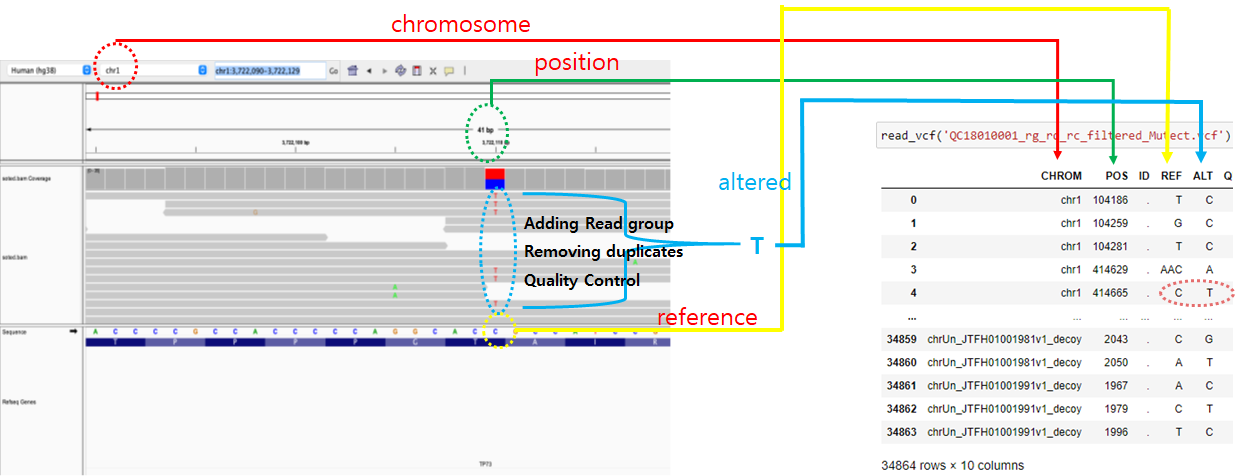
- 생성된 bam파일은 binary 정보를 담고 있으므로 육안으로 파악할 수 없음 -> IGV viewer를 이용하여 확인 가능

- reference 파일과 동일한 위치에 있는 sample 파일(bam)의 read 정보를 통해 변이를 확인할 수 있음
5. Variant Calling with Filtering
- bam 파일에 있는 read를 통합하고, 중복 정보를 제거하여 통계적으로 타당한 변이 정보를 남겨 base quality를 보정
- 이렇게 해서 최종 variant calling 결과가 남게 되고,
- 통계적으로 미미한 정보를 filtering한 결과를 VCF 파일로 저장하게 됨
- bam 파일과 vcf 파일의 정보를 비교하면 다음과 같음
6. Annotation
- VCF 파일에서는 변이 정보를 확인할 수 있으나, 변이의 의미는 알 수 없음
- 변이의 성질, 기능 및 impact 정도는 snpEFF라는 툴로 확인해 볼 수 있음 (local)
- 또는 wANNOVAR나 ENSEMBL에 vcf 파일을 업로드하면 분석된 정보를 확인할 수 있음 (web)

elsenbl에 업로드하여 확인되는 변이 영역과 종류

wANNOVAR에 업로드하여 받게되는 분석 테이블
7. Quality Score
- fastQC 등을 이용하여 전체적 데이터의 퀄리티 체크

- 전체적인 sequence quality, quality score 및 감소된 duplicates를 확인할 수 있음
'데이터 분석 > 생물 및 의료 데이터' 카테고리의 다른 글
| [유전체] Genetic Risk Score (GRS) 계산 (0) | 2023.01.05 |
|---|---|
| 시계열 데이터 (0) | 2023.01.04 |
| [유전체] genotype 별 SNP sample 추출 (0) | 2023.01.04 |
| [유전체] plink에서 sample 그룹 추출 (0) | 2023.01.04 |
| [유전체] Imputation (0) | 2023.01.04 |
- Total
- Today
- Yesterday
- pmm
- 그룹비교
- SNP
- 생존분석
- featuremap
- sequenced data
- missing_value
- cnn
- PTB
- 인공지능
- pre-train
- r
- gray2rgb
- HRV
- GPU설치
- plink
- NGS
- 평균분석
- 생존함수
- sounddevice
- fasta
- psychopy
- vcf
- 생존곡선
- 딥러닝
- 실험통계
- rgb2gray
- Bioinfo
- GradCam
- ECG
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| 27 | 28 | 29 | 30 |